


德勤概述欧盟监管变革及其对全球生命科学行业的影响
引言:随着欧盟药品立法的加强,欧洲药品管理局(EMA)的职权范围不断扩大。目前,针对孤儿药、草药、儿科药品和先进治疗医药产品的审评与管理,EMA都具备专门的法规,并不断推陈出新。近年来,为聚焦公众健康需求,保障监管信息透明度,鼓励病患积极参与到监管工作中,完善药品安全性与有效性的实时监测,EMA先后制定、更新并发布了新版的欧盟《药物警戒法规》《反伪造药品法令》《临床试验法规》《医疗器械法规》等法规与法令,以适应当下欧洲医药产业发展升级和监管环境发生的变化,并对全球的生命科学行业产生了持续性的影响。6月,德勤会计师事务所(Deloitte)发布题为《大有所图:欧盟监管变革及其对全球生命科学行业的影响》报告[1],概述了近期欧盟监管法规的持续性变化,并提出生命科学行业企业应主动了解法规动向,理解监管政策变化对企业业务产生的影响。
一、背景概述
生命科学行业是全世界最为规范的行业之一。随着监管法规日益严格,监管内容日益详尽,各企业需实施更加严格的合规政策,促进跨部门协作,加强数据管理,提升数据完整性。欧盟近期持续开展的监管改革预期将对各企业产生极大的影响。目前在欧盟范围内销售产品的制药、生物技术或医疗技术企业将受到一系列监管改革的影响,如药品鉴定标准、更加完善的药物不良反应系统(EudraVigilance System)、最新的《医疗器械法规》和《临床试验法规》,以及《反伪造药品法令》等[2]。
二、近期欧盟监管法规的变化
纵观EMA公布的新版法规,可以看出欧盟针对医药产品的管理框架正不断迈向协作化、统一化、创新化、精简化与可追溯化。在报告中,德勤会计师事务所从9个方面概述了欧盟近期的监管政策变化(图1)。
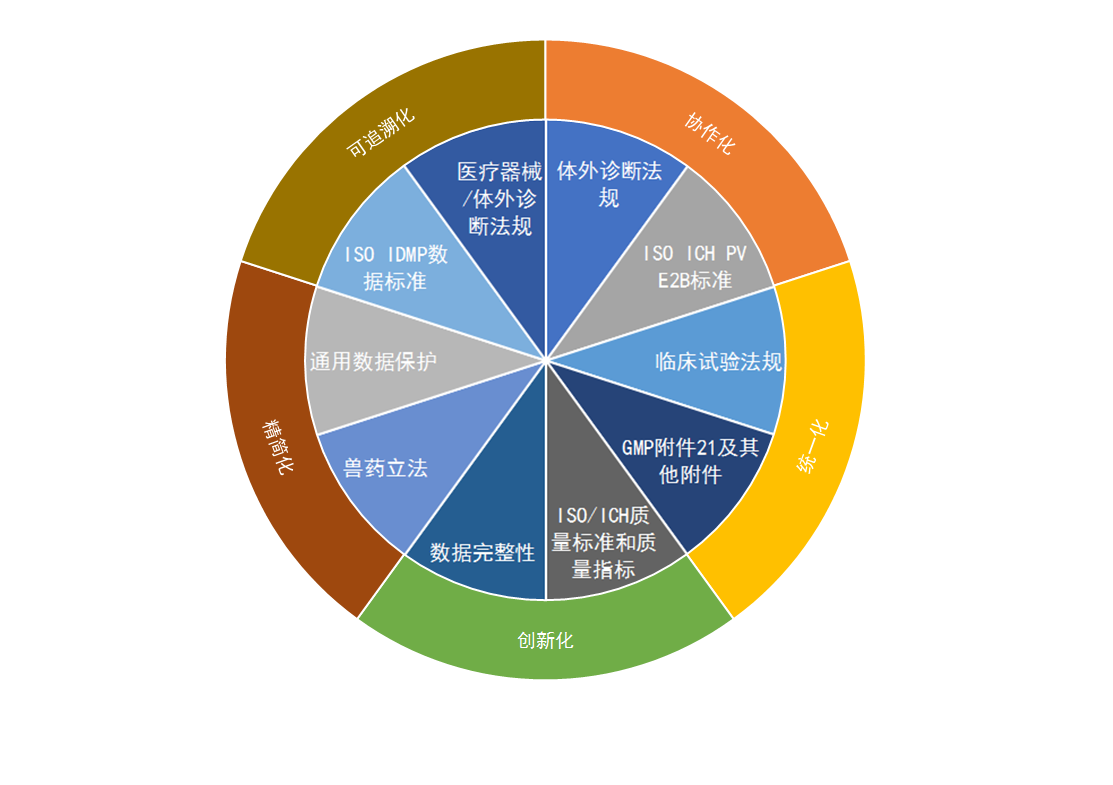
图1 欧盟的监管政策变化概览
1、药品鉴定(IDMP)数据标准
IDMP数据标准是为了响应全球药品协调规范要求,由国际标准化组织、各国家或地区的主管机构、行业协会及其他相关团体所开发并执行的一系列全球数据标准。IDMP标准要求制药企业通过电子方式上传详细的产品数据,并持续更新相关内容。该标准将有助于加快全球药品字典和产品档案的建立;将全球监管机构的产品与安全信息相关联;增加行业的产品危险信号检测能力,从而快速识别产品风险和产品问题,并实施产品召回;并将关键产品信息导入医疗保健系统中。
2、《医疗器械法规》(MDR)
新版的MDR有六大革命性变化,包括提升法律强制性级别、合理划分职责权限、扩大产品覆盖范围、大幅提升临床评估要求、严格审批高风险产品、强化追溯和监督系统。重大变化具体包括重新划分第三类医疗器械范畴、加强临床评估与安全性监测,以及变更技术文件格式等。
3、《临床试验法规》
欧盟《临床试验法规》规定,EMA需要提供、更新并维护信息技术平台系统,以改善欧洲临床试验的申请、评估与监测。该法规的精简化体现在以下方面:简化评估/授权临床试验的程序,在启动新临床研究的过程中减少重复性工作并减少时间延误;由于已授权药物再次开展临床试验与一般临床试验相比风险较小,因此针对已授权药物所进行的试验引入较宽松的监管制度;简化报告要求,避免研究人员将相同的信息重复提交给多个机构;正式承认共同资助,试验可以由多个组织牵头;引入临床试验单一决策的概念,减轻成员国的行政负担。
4、更加完善的药物不良反应系统
更新后的欧盟《药物警戒法规》要求EMA加强药物不良反应数据库的建设,以提供更加精简的报告和更高质量的数据,并改善检索、分析及追溯功能;此外,还要与ICH(ISO人用药品注册技术规定国际协调会议)的个案病例安全报告(ICSR)新版标准接轨。
5、《反伪造药品法令》(FMD)
根据最新的FMD,所有出口到欧盟地区的原料药均需出具出口国监管部门的书面声明,证明其符合相当于欧盟GMP(药品生产质量管理)规范的要求;同时,保证对生产企业实施定期而严格的GMP突击检查,一旦发现违规,立即采取措施;此外,必须及时向欧方通报违反欧盟GMP的案例。或者也可选择申请被列入一份“第三国”名单,以表明申请国与欧盟具有等同性的监督和检测体系。
6、药品生命周期管理的技术和法规考虑(ICH Q12)
ICH Q12将分阶段进行,通过提高效率、确保后续产品的持续供给来改善批准后变更的过程;改善知识管理系统;确定管理和提交批准后变更的标准。ICH Q12将带来如下利好因素:通过产品生命周期中的CMC变更管理流程来提高药品供应的可靠性;监管档案更加标准规范,更加卓有成效;增强对批准后变更管理协议的监管工具的使用;提高产品制造效率,完善持续的制造过程;减少产品的变异性;以及基于风险开展监管支撑。
7、欧盟药品生产质量管理规范附件21-药品进口规范
欧盟GMP新附件21旨在解决由多国制造并进口到欧盟的药品需要多个许可证的问题。其主要目标是为药品进口商提供GMP要求的额外指导,并明确这些要求适用于参与进口活动的不同实体(包括制造商、销售商、代理商等)的情况。尽管欧盟GMP新附件21尚未定稿,但根据其内容和指导精神,企业应评估某项药品的生产若涉及多个产地,是否需要多个许可。
8、产品制造的质量指标
美国食品与药品管理局(FDA)发布的《FDA质量量度技术符合性指南》概述了行业应向FDA提交的建议数据,并清晰描述了FDA对所要求质量量度的期望。此外,FDA定位于质量文化,还提出了一组可选参数的建议,包括高级管理层参与指标、措施和预防措施效能指标,以及过程能力或性能指标。
9、兽药产品(VMP)提案
关于兽药产品的立法提案旨在增加欧盟VMP的可用性,同时减少行政负担,解决耐药性(AMR)的风险。主要内容包括:对制造、进口或出口VMP予以授权时,将要求审查VMP的中间产品、赋形剂及其活性物质;延长大多数物种的技术数据保护期;要求顺势疗法VMPs注册,并使其符合新标准等。
三、生命科学企业的应对措施
欧洲近期的法规变化将引起全球生命科学行业重大变革。更新后的欧洲法规将推动生命科学公司企业进行内部改革,并影响当前的组织架构、管理方式、生产流程及技术研发。面对不断变化的监管要求,建立能够协调统一、及时有效地管理经营模式,同时保证成本效益,成为各企业面临的一项挑战(图2)。
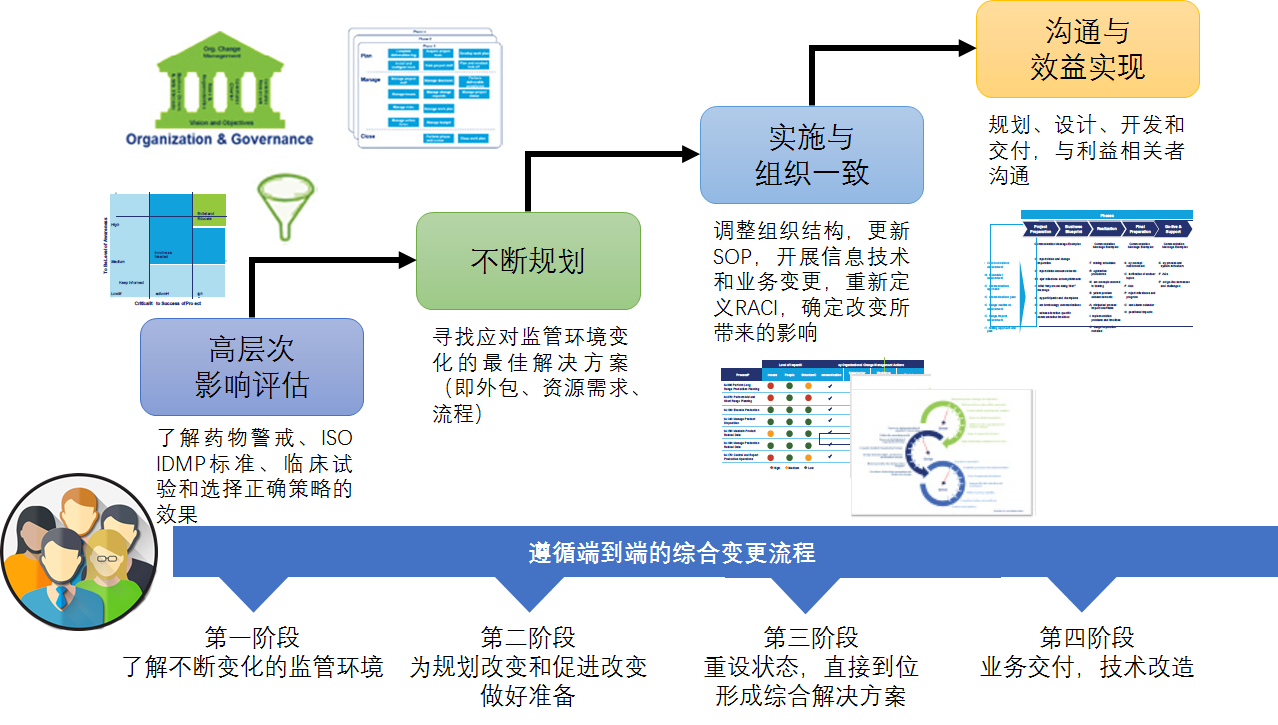
图2 企业应对监管环境变化的管理与掌控过程
欧盟生命科学领域相关法规的变化正在提升知识化水平或知识化程度,规范、简化和统一相关的法规与程序,并提高制造商、分销商、服务提供商、第三方指定认证机构和成员国的效率与可靠性,并将保护患者的人身安全、改善患者的身体状况作为最终目标。 (李祯祺 王玥)
[1] The bigger picture:Impact of EU regulatory change on the global life sciences industry. https://www2.deloitte.com/content/dam/Deloitte/global/Documents/Life-Sciences-Health-Care/gx-lshc-changing-eu-regulatory-landscape.pdf
[2] 2017 Global life sciences outlook. https://www2.deloitte.com/content/dam/Deloitte/global/Documents/Life-Sciences-Health-Care/gx-lshc-2017-life-sciences-outlook.pdf